
In the summer of 2020, biology labs around the world were quiet. Centrifuges and microscopes gathered dust in the pandemic stillness.
But in a makeshift lab in her home basement, UC Santa Cruz genomics researcher Karen Miga was busier than ever.
She was co-leading a vibrant and diverse community of scientists from around the world, in an attempt to sequence the first fully complete human genome. The researchers—ranging from students to seasoned scientists—hoped to fill in gaps that had been impossible to make sense of when the first draft of the human genome was revealed in 2000.

Adam Phillippy of the National Human Genome Research Institute (left) and UC Santa Cruz genomics researcher Karen Miga, at the T2T Consortium’s recent two-day conference at UC Santa Cruz. (Photo by Carolyn Lagattuta)
Miga, like her collaborators, had no funding to support the project, so she squeezed the work into spare moments, surrounded by baby toys to distract her young kids. When they finally fell asleep each night, Miga peered at her computer for hours, poring over long strings of letters that spell out human DNA sequences. In the early morning before her family awoke, she did the same.
“This was very much my passion project,” said Miga, who is now an assistant professor of biomolecular engineering. “I think a lot of us could actually carve out more headspace for this than we would have otherwise been able to, because during that phase of the pandemic we weren’t teaching, we weren’t traveling.”
The team of scientists was known as the Telomere-to-Telomere (T2T) Consortium, named for the protective caps, or telomeres, on the ends of chromosomes. By the end of that summer, the consortium’s work had paid off; they had fully sequenced all 23 different chromosomes contained in human cells, from telomere to telomere, revealing parts of the genome for the first time ever. The new sequences have implications for understanding genetic diseases, human diversity, and evolution.
In March 2022, the consortium published the final results in the journal Science, garnering them sudden attention and accolades—including a spot for Miga on Time magazine’s list of the 100 most influential people of 2022. Even after her red carpet walk at the Time 100 Gala in New York City, Miga remains humble about the project, crediting the team as a whole and focusing on the importance of the findings for future research.
“I think it took both the scientific community and the public very much by surprise,” said Distinguished Professor of Biomolecular Engineering David Haussler, director of the UC Santa Cruz Genomics Institute. “Karen and her colleagues undertook this project quietly, without grandstanding about it, just because they loved science and wanted to better understand the genome.”
Holes in the genome
The genome contained in human cells—everything that dictates who we are biologically and how our molecules function—is made up of more than 3 billion chemical base pairs of 4 different letters, the building blocks of DNA.
In 1990, scientists had begun to develop new ways of determining the order used by these base pairs to spell out genes, and a concerted effort to sequence an entire human genome was launched. Progress was slow and expensive; the researchers had to develop entirely new technologies to make headway. Haussler and colleagues at UCSC joined the publicly funded effort in 1999, racing against a commercial venture to complete the first sequence.
The way scientists sequence an entire genome involves chopping it up into small bits, sequencing each bit, and then using computer algorithms to piece those strings of letters back together. At the turn of the millennium, technology could only sequence about 1,000 base pairs at once. An entire chromosome is tens of millions to hundreds of millions of base pairs long, so that meant lots of pieces, or “reads,” per chromosome.
“You can only read a tiny bit of the genome at a time, and you don’t know exactly which part you’re reading,” said Haussler. “Putting it all together is like solving a very complex jigsaw puzzle.”
Parts of this puzzle were relatively simple to assemble. But other sections of the genome contain vast regions of repetitive DNA. Like assembling the solid blue sky of a jigsaw puzzle, figuring out how these pieces are arranged is difficult. In 2000, when Haussler’s team launched the UCSC Genome Browser to make the first draft human genome accessible to anyone, around 200 million base pairs were missing—almost 8% of the genome. The technology of the time wasn’t able to account for these pairs.
“We had to leave out these sections of the genome,” said Haussler. “We just simply could not tell you what the exact sequences were in all these places.”
Obsessed with satellite DNA
The summer that the 92%-complete human genome went live on the UCSC Genome Browser, Miga had just finished her bachelor’s degree at the University of Tennessee. Already, she was enamored with genomics, and she would soon join a lab studying the repetitive sequences of DNA that had tripped up Haussler and his colleagues.
At the time, some scientists called these and other types of repeating sections “junk DNA,” implying that they had little function. Many repeating sections were less dense than the rest of the genome, floating on top when DNA was separated by density, and hence were called “satellite DNA.” Miga, and many of the researchers she worked with as she completed her graduate degrees—first a master’s at Case Western Reserve University and then a doctorate at Duke University—thought that satellite DNA was more than just junk.
“These parts of the genome are fundamental to life; if you remove them, cells and organisms don’t survive,” said Miga. “That doesn’t sound like junk.”
As she brainstormed what line of research she wanted to pursue, she kept coming back to questions about satellite DNA and its role in biology and human health.
“There was this big black box around satellite DNA, and I could see, for years and years, that there would be endless fascinating questions to keep answering,” she said. “Studying these regions felt like being an explorer.”
Over the coming years, Miga’s self-described obsession with satellite DNA continued, despite the fact that these regions still weren’t sequenced. For her graduate research, she homed in on one particular type of satellite DNA, located at the very middle of each chromosome in an area called the centromere. Then, in 2011, she began talking to Haussler about coming to UCSC for a postdoctoral fellowship.
“She told me right off the bat that she wanted to sequence and study the satellite regions of the genome; that she thought they were important regions in genetics even though everyone else was ignoring them,” remembered Haussler. “I told her she could go for it, pursue this dream, but that she would be mostly on her own—my lab was working on trying to understand all kinds of other things about the genome.”
Filling in the gaps
In the 15 years after the first draft human genome, gene-sequencing technology advanced rapidly. Scientists, however, continued to piece together the same-size bits of DNA (albeit faster and more cheaply), so the challenge of sequencing satellite DNA didn’t lessen at first.
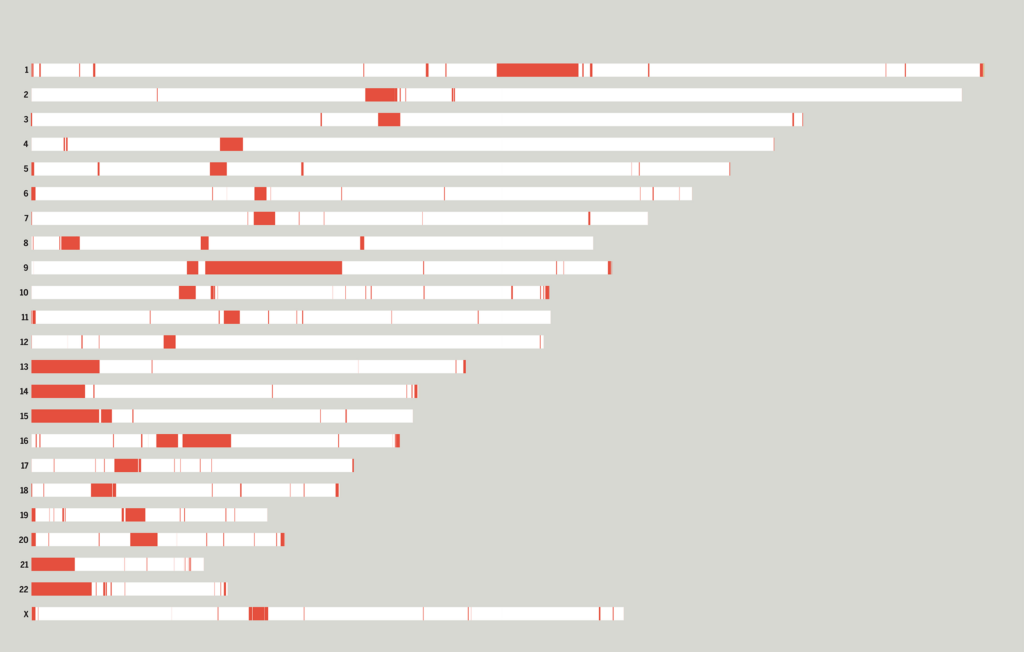
A diagram that shows the new segments in each chromosome.
That all changed about five years ago, when researchers pioneered ultra-long-read sequencing. Instead of dividing DNA into 1,000 base-pair segments, these technologies—including nanopore sequencing, spearheaded by a team at UCSC—let them sequence up to a million base pairs in one single go.
If the genome had been a thousand-piece puzzle with hard-to-assemble splotches of blue sky, now it was a 25-piece puzzle, where sky (or repetitive DNA) was lumped together on pieces with identifiable features (such as known genes, or other previously characterized sequences flanking the gap). Suddenly, Miga’s goal of sequencing the satellite regions in the genome seemed doable.
“As soon as I saw there was a way to sequence reads of a hundred thousand bases or more, I knew it was time to complete the human genome,” said Miga.
She teamed up with Adam Phillippy of the National Human Genome Research Institute, a computational biologist with expertise in genome assembly methods, who had a shared interest in sequencing tricky stretches of DNA. To test whether their goal of sequencing the whole genome was feasible, they agreed to begin with the X chromosome. It worked; in 2019 at a preeminent genome meeting , they presented a completed X chromosome. In its center, they managed to assemble the sequence of a massive, 3-million-base-pair, highly repetitive centromere—exactly the kind of thing Miga had set out to do.
A kaleidoscope of talent
Miga and Phillippy could have kept working on the rest of the genome themselves, or with a few close collaborators. Most academic research gets done that way: behind closed doors. Instead—at the same presentation where they debuted the X chromosome—they launched the T2T Consortium, complete with a public website where their data would be immediately posted as they generated it.
“I always thought as a researcher, I work quietly in my little corner of the field and I generate data and then I publish it,” said Miga. “I was this early-career researcher with very little job security, and I had a lot to risk by putting all our data out there right from the beginning.”
At the time, Miga was an assistant research scientist at UCSC—a temporary position while she searched for faculty jobs. Being a member of a consortium doesn’t usually look as good on an academic job application as spearheading a project by yourself. But despite the risks, Miga and Phillippy reasoned that more people tackling the gaps in the genome would make the project more doable, and move more quickly. It also fit closely within the existing ethos of the UC Santa Cruz Genomics Institute.
“When we posted the first draft of the human genome on the internet [in 2000] with no intellectual property claimed at all, it was a statement about what Santa Cruz believed—and still believes—about open data. Anyone should be able to participate in science, share data, and move toward a greater understanding of our world and our universe,” Miga said.
Over the next few months, interested scientists flocked to their website, both to join the consortium in an official way, or to peruse data and contribute comments. Eventually, the group approached 100 scientists.
“It became a kaleidoscope of talent,” said Miga.
In one case, a student working with Evan Eichler—Miga’s mentor from Case Western, now at the University of Washington—noticed an error and helped fix it. His observation led to new ways of verifying the gene sequences and improved the project, Miga said. In other instances, researchers took on whole sections of a chromosome to analyze in their spare time. Associate Professor of Biomolecular Engineering Benedict Paten, who has the office next door to Miga at UCSC, joined the consortium and spearheaded efforts to identify genes contained in the newly sequenced DNA.
“We never would have developed this type of vibrant, collaborative community if we hadn’t released our data so openly,” said Miga. “So I think that was a big lesson for me, personally, about the benefit of open-data science and sharing.”
By the time this community had assembled, COVID-19 was circulating around the world. With the new ability to work remotely, Miga moved from Santa Cruz to Seattle, where her husband’s job was based. Others on the team found new pockets of time to devote to the project, which began moving even faster than expected.
More on Nanopore Sequencing Innovation at UCSC
From concept to commercialization: How UCSC researchers revolutionized DNA sequencing
More than a quarter century since the first patents were filed, the UCSC researchers who pioneered nanopore sequencing reflect on the impact of their invention. Read more>>
In late May of 2020, one of Adam Phillippy’s postdoctoral fellows, Sergey Nurk, shared his first string graph, which shows the overlaps between sequenced bits of DNA and is the first step toward spelling out a final chromosome sequence. For the first time, Miga and Phillippy saw—right there on their computer screens—that it would be possible to sequence the entire human genome. That pandemic summer, they and their collaborators put their heads down and worked, from home offices and basements around the globe, to complete the project. Preparing the results for publishing took until the following spring, when the news broke across the world.
Capturing human genetic diversity
While Miga’s nights that summer were spent on the T2T Consortium, her days were spent on something else: the Human Pangenome Reference Consortium. This project, which Miga was launching through her role at the UC Santa Cruz Genomics Institute, aims to fully sequence a thousand genomes from a diverse set of people to capture human genetic diversity. The two efforts fit closely together—high-quality complete genomes are part of the goal of the Pangenome Consortium, which is also run by Paten and Haussler.
“Having the ability to sequence an entire genome, telomere to telomere, is an extremely important ingredient in putting together the pangenome,” said Paten.
Complete genomes—both the first T2T genome published this year, and the eventual set of genomes included in the pangenome—will likely reveal new, previously hidden aspects of how the DNA in human cells dictates biology and disease. Already, the scientists identified nearly 2,000 potential new genes embedded in the newly completed sequences.
“I think we’ll see a lot of new scientific knowledge coming out of this,” said Haussler. “We’ll be discovering things about the biology of a cell, and about disease genes, left and right.”
For their part, Miga and Paten are proud of the potential implications of the T2T project for shedding new light on disease and human biology. But some of the pride in finishing the first telomere-to-telomere, gapless human genome is also more basic. They showed that it could be done. They solved a puzzle.
“There is definitely a nerdy side to this,” said Paten. “There were a small number of very technical genomics groups out there who just wanted the ‘geek cred’ of being able to complete this incredibly challenging genome.”
What they discovered is that geek cred doesn’t have to come from competition, but from working together. As for Miga, her risk-taking paid off. Last summer she was named an assistant professor at UCSC and, in August 2021, she moved back to Santa Cruz with her family.
“I am very proud of our collective efforts,” said Miga. “It’s true there were a bunch of genome geeks behind this work, but there is something personal about the announcement of the first complete view of a human genome. It is more than a string of letters; it is new information about our humanity.”
Sarah C. P. Williams, a graduate of the UC Santa Cruz Science Communication Program, is an award-winning science writer who explores the stories behind scientific inquiry and discovery for institutions and publications around the world.